

UCD is a rare disease affecting an estimated 1 in 35,000 live births in the United States, though actual incidence may be greater.4,5,7-9
About UCDs
Urea Cycle Disorder Pathophysiology and Prevalence
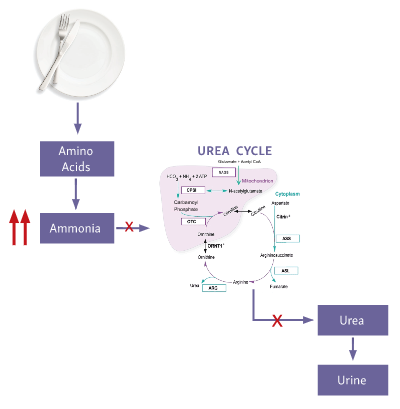
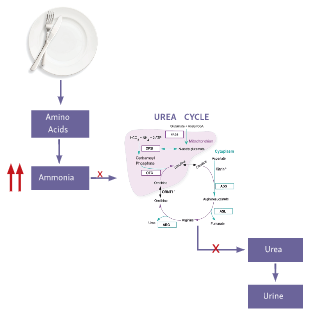
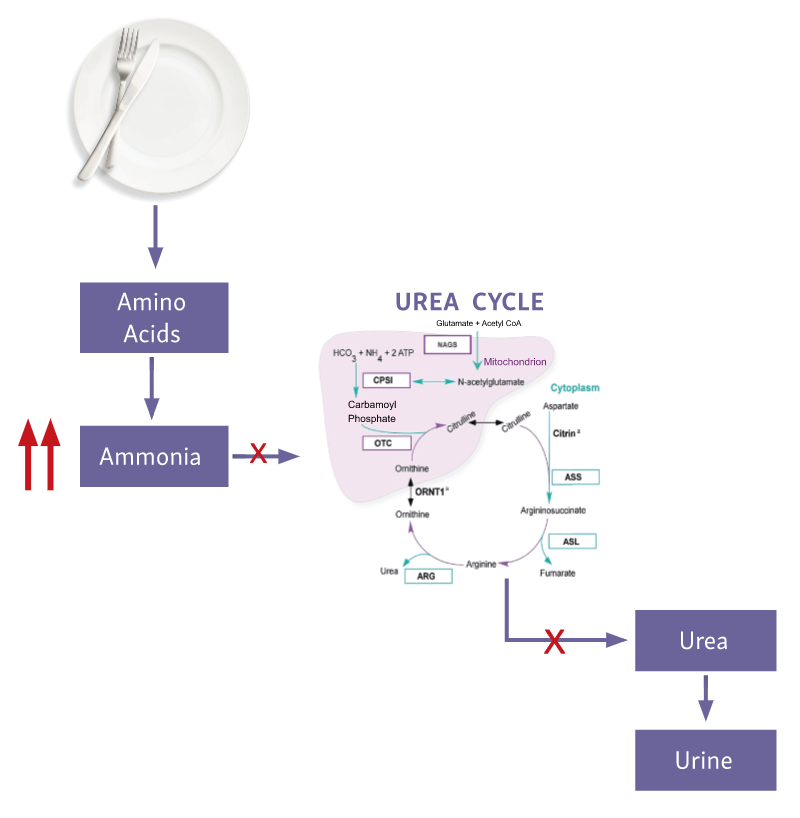
A UCD affects the process of protein degradation in the body.1,2 When protein is broken down into amino acids, any excess nitrogen is converted into ammonia, which is then converted by the urea cycle into urea for safe removal from the body.2,3
An enzyme or transporter that is missing or defective in the urea cycle may cause a UCD.4 The urea cycle is, therefore, unable to excrete toxic ammonia resulting in a buildup of ammonia in the blood.3
The elevated levels of ammonia in the body can cause fatigue, confusion, headaches, vomiting, behavioral changes, learning difficulties, brain damage, or coma.2,5 Presentation of these symptoms can range from asymptomatic to severe depending on the affected enzyme and the extent of the enzyme deficiency.4,6
Urea Cycle Disorder Inheritance
Eight UCD subtypes are identified based on the affected enzyme or transporter3,10:
- Ornithine transcarbamylase (OTC) deficiency
- Carbamoyl phosphate synthetase 1 (CPS1) deficiency
- Argininosuccinate synthetase (ASS) deficiency or citrullinemia type 1 (CTLN1)
- Argininosuccinate lyase (ASL) deficiency
- Arginase-1 (ARG) deficiency
- N-acetylglutamate synthase (NAGS) deficiency
- Hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome
- Citrin deficiency or citrullinemia type II (CTLN2)
OTC deficiency is the only X-linked UCD subtype. A mother may pass the affected OTC gene to her sons or daughters, while a father may pass the affected OTC gene to his daughters.4 Individuals with OTC deficiency may experience symptoms and require treatment.4,5,11
All other UCD subtypes are inherited in an autosomal recessive manner.4 The presentation and severity of symptoms in individuals with an autosomal recessive UCD may vary based on the specific UCD subtype.4
Rarely, a UCD is the result of a new—or de novo—mutation.4 These mutations, similar to inherited mutations, may be passed on to children.4






Urea Cycle Disorder Signs & Symptoms
Signs and symptoms of UCD can vary person to person.
Physical symptoms5:
- Fatigue
- Headaches
- Seizures
- Vomiting
- Poor appetite/protein aversion
- Slow growth
- Vision loss
- Ataxia
- Muscle weakness
- Tremor
Cognitive/behavioral symptoms5,13,14:
- Mood swings
- Attention problems
- Learning disabilities
- Hallucinations
- Personality changes
- Aggression
The wide range of nonspecific symptoms makes identifying a UCD challenging. If a UCD is suspected, a specialist may be able to help make a diagnosis.
Connect Your Patient With a Provider Experienced in Diagnosing and Treating UCDs
Other clinical signs of UCD include high ammonia levels and elevated glutamine, urinary orotic acid, citrulline, and alanine aminotransferase biomarkers. High ammonia levels can result in a wide range of effects including hyperammonemic encephalopathy, which can lead to coma and death.
Effects of High Ammonia Levels


High Ammonia Levels Can Cause Neurological Deficits
Elevated ammonia levels, whether acute or chronic, may be toxic and have significant neurological consequences.16
With acute or peak ammonia elevations, increases in blood ammonia concentration may result in cerebral edema, coma, and eventual death.1,8 These risks of mortality and neurological damage are influenced by the number, extent, and duration of acute episodes of a hyperammonemic crisis.17-25
Similarly, chronic or moderate ammonia elevations over time may result in negative neurological and cognitive effects.4,8,16,27 The risk of irreversible brain damage is not predicted by the presence or absence of symptoms related to elevated ammonia levels.4,8,16,27 Even moderate increases in blood ammonia concentration may also lead to coma or death if left untreated.8


Potential triggers of hyperammonemic crises include illness, poor diet, medication noncompliance, and stressful life events.9
The first hyperammonemic crisis may have severe neurological outcomes of disability or death when ammonia levels are particularly elevated. Neurocognitive function further declines with subsequent hyperammonemic events.
The Specific Enzyme Deficiency Determines the UCD Type